

























Members: Tim Lee, Xinchuan Huang, Partha Bera
Objectives:
- Compute highly accurate molecular Infra-Red (IR) rovibrational line lists for molecules in the Interstellar Medium (ISM), and planetary and exoplanetary atmospheres, reliable up to ~ 2000 – 3000 K
- Study and characterize the evolution of complex organic molecules in the gas phase under astrophysical conditions by identifying the reaction mechanisms of formation and destruction of complex organic molecules
- Compute accurate rovibrational spectroscopic constants for small molecules under astrophysical conditions, including simulated spectra, and in some cases low-temperature line lists
- Determine accurate anharmonic spectra and fully anharmonic cascade emission spectra for complex organics, including Polycyclic Aromatic Hydrocarbon (PAH) molecules
- Together with experimentalists, explore condensed-phase reaction mechanisms for molecular evolution of complex organics in astrophysical ices leading to molecules or precursors of interest in astrobiology, including nucleic acids, sugars, nitrogenated ring compounds, etc.
Expertise/Experience
We have over three decades of experience in the development of ab initio quantum chemistry methods. Some examples include:
- Development of robust composite schemes for computing highly accurate quartic force fields (QFFs) and potential energy surfaces (PESs)
- Development of an efficient open-shell perturbation theory for restricted Hartree-Fock reference functions
- Development of the first production version program of the CCSD(T) coupled-cluster theory, considered to be the gold standard (or the best compromise between quality and cost) in quantum chemistry
- Development of the first and most efficient CCSD(T) analytical gradient method
- Development of the T1 diagnostic in coupled-cluster theory
We have many decades of experience in the application of ab initio and density functional theory quantum chemistry methods to solve a wide variety of chemical and spectroscopic problems related to astrochemistry and atmospheric chemistry. Some examples include:
- Electronic structure of atoms and molecules, electronic state energies, excitation energies, ionization potentials, electron affinities, molecular orbitals
- Accurate molecular structures, dipole moments, polarizabilities
- Electronic (UV-Visible) spectra, valence, charge-transfer spectra, CIS, EOM-CCSD, TDDFT
- Pure rotational (microwave) spectra; Vibrational infrared (IR) spectra
- Rovibrational spectra, Vibrational anharmonicities, Quartic force fields
- Potential energy surfaces, Dipole moment surfaces, Molecular line-lists, Opacities
- Reaction enthalpies, reaction kinetics, rate constants, reaction pathways, branching ratios, transition states
- Global Warming Potentials of greenhouse gases, Rovibrational spectra of potential biosignature, and technosignature gases
- Reaction mechanism exploration studies involving neutral, radical, and ionic molecules in the gas-phase as well as ice phases; Ab initio molecular dynamics
Facilities
Resources
- Access to the Pleiades Supercomputer of the High-End Computing Capability of the NASA Advanced Supercomputer (NAS) division of NASA
- 14 node in-house cluster that includes servers and blades for running long-time and expensive computations that need weeks and months
Access to Ab initio software packages
- Q-Chem: Q-Chem is a comprehensive ab initio quantum chemistry software for accurate predictions of molecular structures, reactivities, and vibrational, electronic and NMR spectra, designed and maintained by M. Head-Gordon and P. Gill, with contributions from many other authors.
- MOLPRO: MOLPRO is a comprehensive system of ab initio programs for advanced molecular electronic structure calculations, designed and maintained by H.-J. Werner and P. J. Knowles, with contributions from many other authors.
- PSI4: PSI4 is an open-source suite of ab initio quantum chemistry programs designed for efficient, high-accuracy simulations of a variety of molecular properties.
- TITAN: TITAN is a set of ab initio programs that predominately include the CCSD(T) method, analytical gradients, and electronic excitation energies, and was developed by T. J. Lee, J. E. Rice, and A. P. Rendell.
- SPECTRO: SPECTRO is a classic vibrational second-order perturbation theory (VPT2) program to compute the rovibrational spectroscopic constants of molecules from a QFF. SPECTRO was developed by J. F. Gaw and N. C. Handy.
- Other programs: VTET is a variational rovibrational program for triatomics and tetraatomics that uses an exact kinetic energy operator, and was developed by D. W. Schwenke; INTDER is a program that will convert QFFs from one type of coordinate system to another (for example, from symmetry internal coordinates to Cartesian coordinates), and was developed by W. D. Allen; PGOPHER is an effective Hamiltonian program that will take rovibrational spectroscopic constants (e.g., from Spectro) and use these to generate a simulated rovibrational spectrum, and was developed by C. M. Western; MULTIMODE is a VSCF-VCI program computing polyatomic rovibrational energies using normal coordinates and a hierarchical n-mode representation which can be applied to a wide class of molecules and molecular complexes, and was developed by S. Carter and J. M. Bowman.

Objective 1. Compute highly accurate molecular Infra-Red (IR) rovibrational line lists for molecules in the Interstellar Medium (ISM), and planetary and exoplanetary atmospheres, reliable up to ~ 2000 – 3000 K
Methodology:
- State-of-the-art ab initio CCSD(T) computations are performed on a grid of points around the global minimum and spectroscopically important regions of a molecule’s PES using a number of electron correlation consistent basis sets that may include f, g, and h as well as diffuse functions; important corrections such as core-correlation, scalar relativistic, and when necessary higher-order electron correlation are included; extrapolation of energies to the one-particle basis set limit is performed; nonadiabatic corrections are estimated
- High-quality potential energy surfaces are fitted from these ab initio energies, and then empirically refined using selected, i.e., the most reliable high-resolution experimental data. The refined PES can provide complete, reliable, and self-consistent predictions for missing bands, weak bands, high energy bands, and/or the bands of other isotopologues
- Together with high-quality ab initio dipole moment surfaces, exact quantum rovibrational and intensity calculations are performed, with or without nonadiabatic corrections, to generate rovibrational IR line lists at both room temperature and in the 1000-3000 K range, with line position accuracy 0.01~0.05 cm-1 and intensity accuracy better than 95-98%.
Applications: NH3, CO2, SO2, N2O, etc. [See the Ames Molecular Spectroscopic Database]
Missions served: SOFIA (EXES, FORECAST, FLITECAM), Venus Express, Herschel Space Telescope (HiFI), Spitzer (IRS), JWST (NIRSpec, MIRI), ALMA
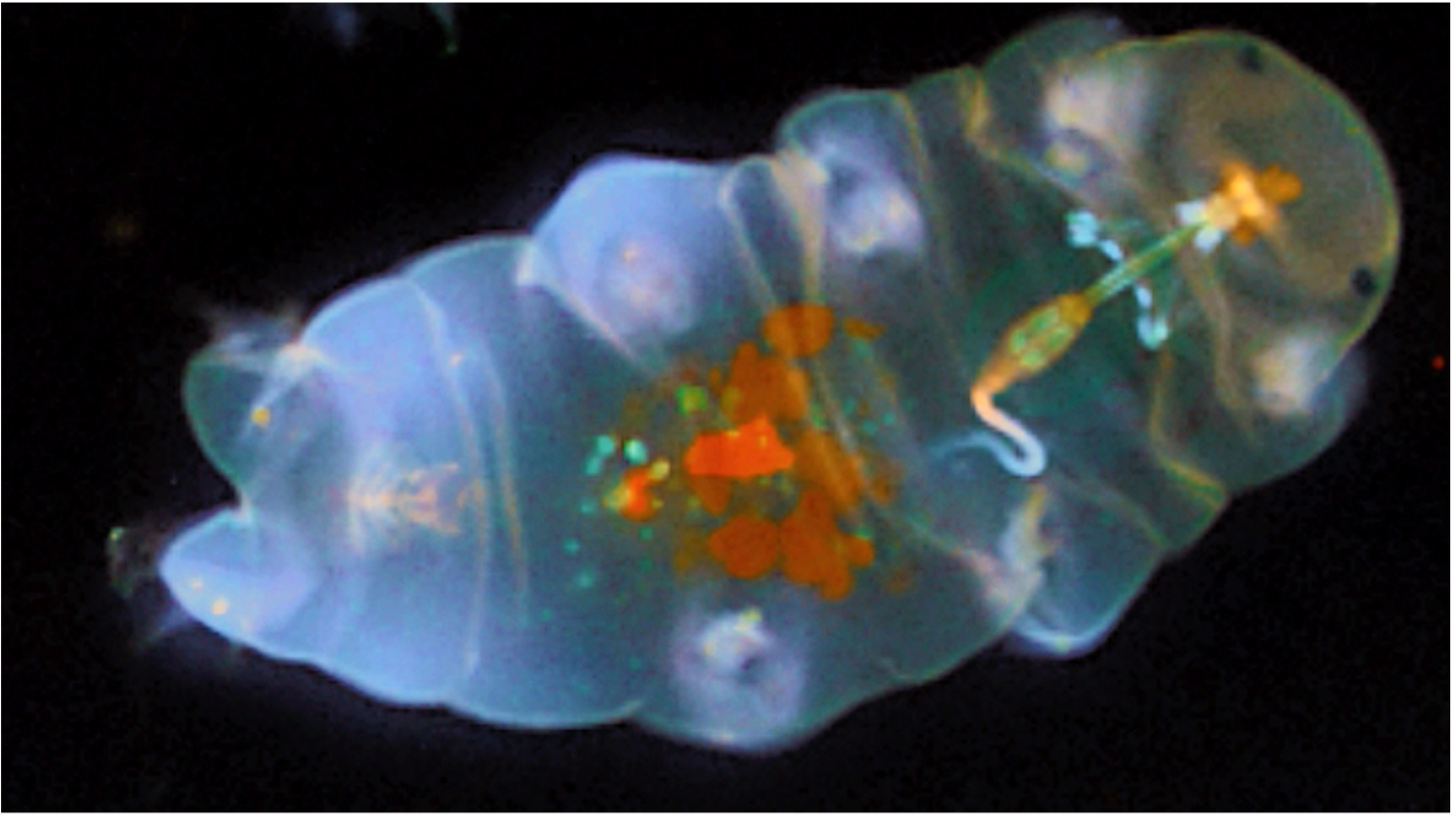
Objective 2. Study and characterize the evolution of complex organic molecules in the gas phase under astrophysical conditions by identifying the reaction mechanisms of formation and destruction of complex organic molecules
Methodologies:
- Bottom-up synthesis of complex organic molecules involving neutral, radical and ionic pathways
- Suitable building blocks (e.g. acetylene) are identified; their reactions with other organics are studied using Quantum Mechanical (QM) methods e.g. DFT, MP2, CCSD(T), etc.
- Structures, energies, reaction energies, transition states, intermediates, products, branching ratios, reaction rate-constants are calculated, and results compared against available experimental data
- UV-Vis, Microwave, and harmonic vibrational infrared spectra are calculated
- Similarly, destruction processes and reaction pathways of larger carbonaceous molecules for top-down formation and destruction of molecules are calculated
- Ab initio molecular dynamics calculations for gas-phase formation of clusters and reaction products are performed
Applications:
- Formation of complex organic molecules especially pure hydrocarbons, e.g., polycyclic aromatic hydrocarbons (PAHs), and clusters
- Formation of nitrogenous hydrocarbons such as nitrogenated organics, nucleic acids, etc. in the gas phase, which are important to origin of life studies
Missions served: JWST, SOFIA, Spitzer, CASSINI, ALMA
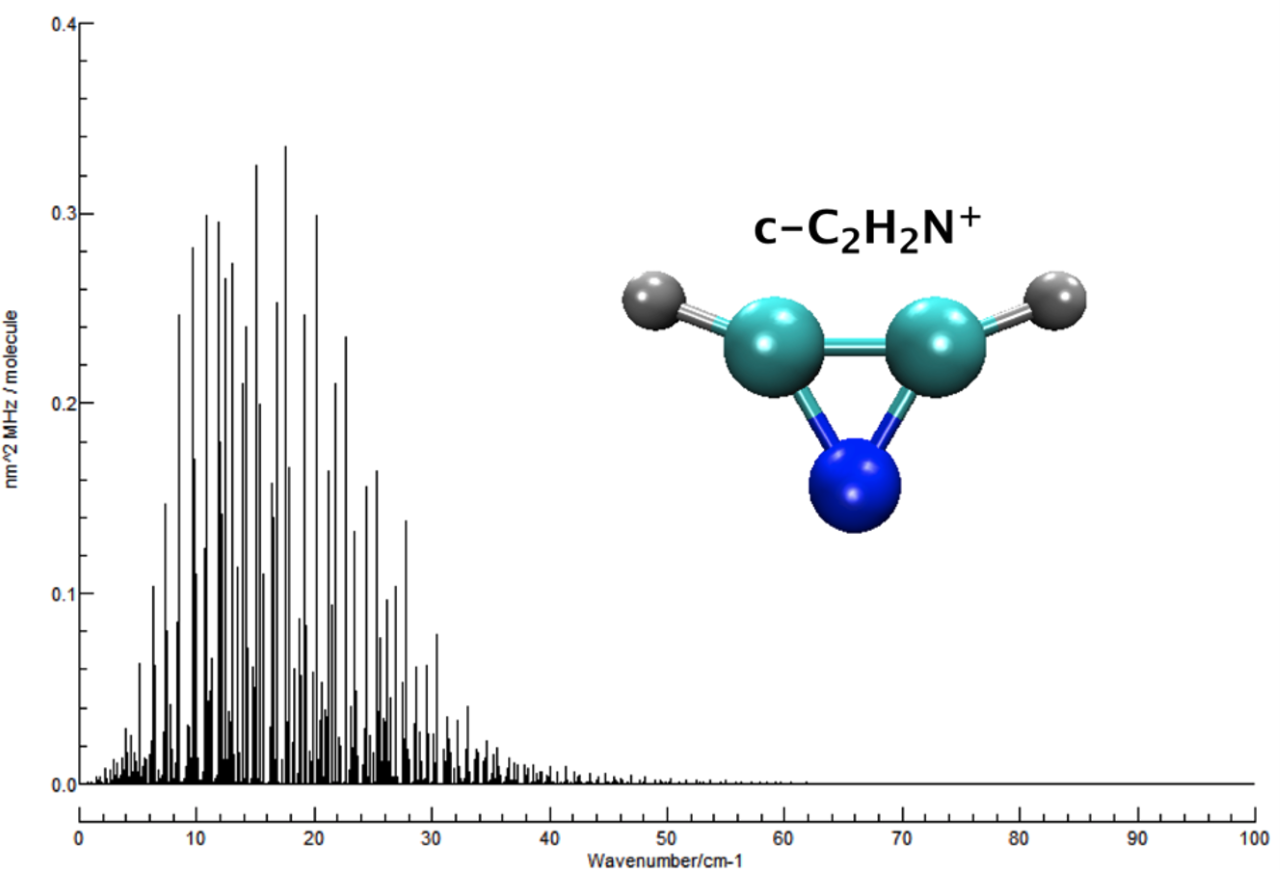
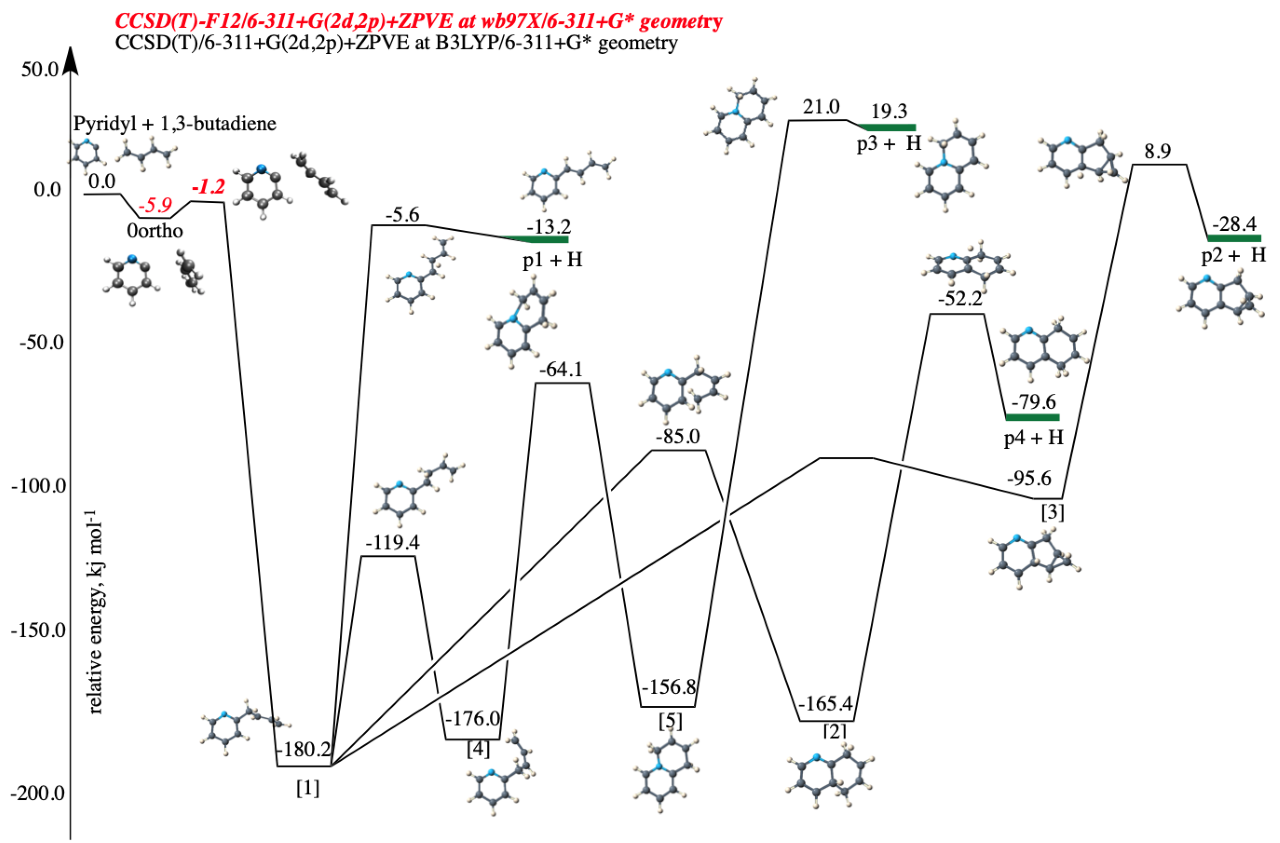
Objective 3. Compute accurate rovibrational spectroscopic constants for small molecules under astrophysical conditions, including simulated spectra, and in some cases low-temperature line lists
Methodology:
- Second-order vibrational perturbation theory (VPT2) analysis is performed on high-quality quartic force fields to obtain reliable anharmonic constants, fundamental vibrational frequencies, rotation-vibration interaction constants, vibrationally averaged rotational constants, quartic and sextic centrifugal distortion constants, Coriolis constants, and anharmonic IR band intensities, band centers for overtone and combination bands; Fermi I, Fermi II, Darling-Dennison, and Coriolis resonances are included, as well as other resonance terms that occur in resonance polyads
- Temperature-dependent IR line lists can be generated from spectroscopic constants, band center information, and anharmonic band intensities using the PGOPHER program; Potential refinements can be done if high-resolution experimental data is available
- For molecules with large amplitude motions, the vibrational configuration interaction (VCI) method is applied
Applications: HO2+, NH2-, CCH-, C3H2, C3H+, CH3OH, N2H+, NO2+, C2H2, C3H3+, HOCO, HC2N, HOCO+, HOCS+, HSCO+, HCN, HCO+, C3H-, N2OH+, C2H3+, HOCS, HSCO, N3+, CNN, HCNN+, CNC, C2H2N+, and many others (see the publication list)
Missions served: SOFIA, Herschel Space Telescope, Spitzer, JWST, Cassini, IRTF, ALMA
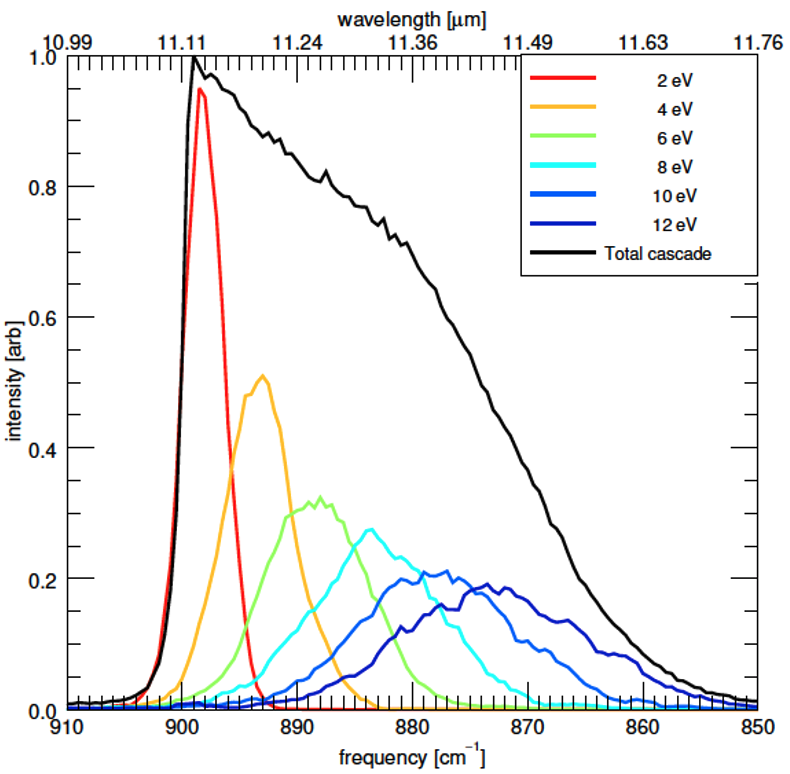
Objective 4. Determine accurate anharmonic spectra and fully anharmonic cascade emission spectra for complex organics, including Polycyclic Aromatic Hydrocarbon (PAH) molecules
Methodology:
- Use DFT to compute a QFF which is then analyzed using VPT2 to compute anharmonic vibrational spectra and anharmonic IR intensities. The molecules are too large to use the more exacting CCSD(T) method, and because of the larger size of the molecules, many Fermi I and II resonances occur and these must be treated as a resonance polyad, which then includes additional coupling terms. The IR intensities of the bands in the polyad are distributed according to the eigenvectors of the polyad. These 0 K spectra are then compared with high-resolution, low-temperature (10 K) experiments to benchmark the DFT method being used.
- Fully anharmonic temperature-dependent spectra are then calculated using the VPT2 anharmonic constants together with a re-diagonalization of the polyad matrices for each energy regime (the only part of the polyad matrix that changes is the diagonal terms).
- The library of temperature-dependent spectra is then used in a biased Monte Carlo walk, known as the Wang-Landau method, to compute fully anharmonic cascade emission spectra of PAH molecules – the exact type of spectra that astronomers observe. These are analyzed and compared to astronomical observations.
Applications: Small to medium-sized PAH molecules
Missions served: Used to assign astronomical spectra relevant for JWST, Spitzer, SOFIA, IRAS, IRTF

Objective 5. Together with experimentalists, explore condensed-phase reaction mechanisms for molecular evolution of complex organics in astrophysical ices leading to molecules or precursors of interest in astrobiology, including nucleic acids, sugars, nitrogenated ring compounds, etc.
Methodology:
- Prebiotic molecules that can be synthesized in astrophysical ices are identified. Ice-phase reaction mechanisms involving neutral, radical, and ionic pathways are explored using methods suitable for condensed phases; Formation and evolution a gamut of important molecules including the ones found in experiments due to the action of UV-radiation in astrophysical ices are investigated.
- Reaction intermediates, products, favored pathways, rate constants, and branching ratios are calculated using ab initio condensed phase methods.
- The results of the ab initio quantum calculations are used to interpret experimental results; extend conclusions beyond experimental results; put constraints on experimental and observational outcomes.
- Calculate useful spectra for intermediates (precursors) and final products.
Applications: Purine, Pyrimidine, Adeinine, Guanine, Thymine, Cytosine, Uracil, Hexamethylenetetramine (HMT), and HMT-derivatives, Amino acids, sugars, other complex organics
Missions served: SOFIA, SPITZER, Stardust, CASSINI, Sample return missions
Find our publications here.